
Water dimer, from ab initio 6-31G** calculation

![]() Water dimer and trimer inside fullerenes
Water dimer and trimer inside fullerenes
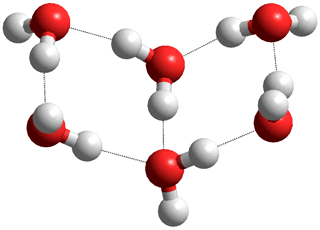
![]() The water trimer, tetramer, and pentamer
The water trimer, tetramer, and pentamer
![]() Small clusters
Small clusters
Much effort has been expended on the structure of small isolated water clusters. The smallest cluster is the dimer. Typically, there is over one water dimer for every thousand free water molecules rising to about one in twenty in steam in the ambient atmosphere. Water dimers contribute significantly to the continuum absorption of atmospheric water vapor, which is important for our warm climate. It absorbs longwave radiation that would otherwise radiate out to space.
A water dimer has one mirror plane of symmetry (CS ) and dimensions from ab initio 6-31G** calculation as shown right. b The charge transfer for this calculation is only 0.03 e− from δ+ve acceptor molecule to δ-ve donor molecule. Its molecular orbitals are shown on another page. A 'high-precision' ab initio method applied on the water dimer showed an O-H bond length of 0.97 Å and H-bond (H···O) length of 1.95 Å (see also above right). A decrease of the O-H-bond length and O···O distance was considered as an uncooperative effect of this single hydrogen bond (O–H···O) [4221].
The variation of water's fugacity coefficient with pressure

The equilibrium constant for dimer formation
2H2O(g) → (H2O)2(g)
is 0.0501 bar−1 at 298.15 K [2052], with dissociation energy of 13.2 kJ ˣ mol−1 [2210a]; (D2O)2 14.9 kJ ˣ mol−1 [2210b] (but see below). The thermochemical properties of the dimer have been determined [2052], and experimental dimer studies have been reviewed [2314].
The dimer formation causes deviations from ideal behavior in gaseous water (see left) [2356].a
The effect of the hydrogen bond on the stretch vibrations is seen below. The activation energy required to switch from one acceptor position to the other on the acceptor molecule (1.88 kJ ˣ mol−1) is only 40% of that required to switch from one donor hydrogen to the other (4.71 kJ ˣ mol−1) on the donor molecule [2314] via the bifurcated state, both of course far lower than the dissociation energy.
| Vibrational bands | symmetric stretch (v1, cm−1) |
bend (v2, cm−1) |
asymmetric stretch (v3, cm−1) |
| H2O monomer [607] | 3657 | 1595 | 3756 |
HO-H···OH2 dimer donor |
3545 [2314] | 1669 [2315] | 3715 [2314] |
| HO-H···OH2 dimer acceptor | 3600 [2314] | 1653 [2315] | 3730 [2314] |
The high-resolution spectra for the out-of-plane librational vibrations, at around 500 cm−1, of the water dimer have been published [2719], as has the rotational spectra around 4 cm−1 [1977b].
Bond energies calculated using 6-31G** basis set

Calculated bond energies for the water dimer are given right using the 6-31G** basis set. Particularly noteworthy is the steepness of the 'wall' inside the optimum position and the more relaxed structures allowed outside the optimum position that still have significant bond energy. The position of the vibrational energy of the hydrogen-bond stretch (≈ 200 cm−1) is shown with the vibrational range of about -0.15 Å +0.3 Å. Although stated in the early literature that the behavior is purely electrostatic at larger distances, this is not true. The most energetically favorable water dimer is shown above right using ab initio calculations with the 6-31G** basis set. It is also shown below with a section through the electron density distribution (high densities around the oxygen atoms have been omitted for clarity). This shows the tetrahedrality of the bonding despite the lack of clearly seen lone pair electrons although a small amount of distortion along the hydrogen bond can be seen. This tetrahedrality is primarily caused by electrostatic effects (that is, repulsion between the positively charged non-bonded hydrogen atoms) rather than the presence of tetrahedrally placed lone pair electrons. The hydrogen-bonded proton has reduced electron density relative to the other protons [222]. Note that, even at temperatures as low as a few kelvin, there are considerable oscillations in the hydrogen bond length (≈ ±0.2 Å at 50 K with timescales ≈ 0.2 ps) and angles (≈ 9 ° at 50 K with timescales ≈ 0.2 ps) [591]. The potential energy surface [1668] and wagging vibration [1743] of the water dimer have been described, as has the energy landscape for a discretized path integral representation of the water dimer [3520].
Water dimer showing the electron density perturbation along the hydrogen bond

Water dimer dimensions
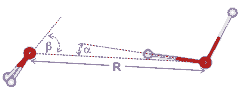
R = 2.976 (+0.000, -0.030) Å, α = 6 ± 20°, β = 57 ± 10° [648]; α is the donor angle and β is the acceptor angle. The dimer (with slightly different geometry) dipole moment is 2.6 D [704]. Although β is close to as expected if the lone pair electrons were placed tetrahedrally (= 109.47°/2), the energy minimum (≈ 21 kJ mol−1) is broad and extends towards β = 0°.
from [3455]
![Water dimer stability from [3455]](images/dimer-stab.gif "Water dimer stability from [3455]")
The relative stability of the dimer dimensions in liquid water has been established for several water models. On the right is shown that for MP2 /aug-cc-pVDZ simulated liquid water [3455]. The energy contours are in the units of kJ ˣ mol−1. The O···O distances (R) and the and the O···O-H angle (α) are as shown in the diagram above right. The minimum is some way from that given by the ab initio 6-31G** calculation of the isolated dimer (given by the small red cross). A typical value range, used for the hydrogen-bond (R < 0.35 nm, α < 30°), includes a large area of high-energy structures.
Dimers of more distant water molecules (≈ 1 nm) show synchronous behavior in their interacting electric fields [2086].
Bond energies of dimers with temperature.
from [2442]
![Bond energies of dimers with temperature. from [2442]](images/dimer-bond-energy.gif "Bond energies of dimers with temperature. from [2442]")
At lower temperatures, the bond energy for (H2O)2 and (D2O)2 dimers reduce with temperature, explained as due to the vibration amplitude growth. However at higher temperatures, the bond energies of both increase with temperature. This phenomenon also occurs in methanol but is difficult to explain [2442].
The intermolecular modes of the water dimer in the frequency region from 70 cm−1 to 550 cm−1 have been investigated using a helium nanodroplet isolation setup [3648]. The full set of 12-dimensional vibrational energy levels of the water dimer ahave been calculated, predicting accurately the experimentally observed intramolecular fundamentals [4449].
There are two HOD dimer isomers, the D-bonded isomer and the H-bonded isomer. The H-bonded isomer ground state energy is 686 J ˣ mol−1 higher than the D-bonded isomer ground state energy [3411].
In place of the hydrogen bonding, it has been proposed to separate the intermolecular interaction energy of water dimers into four physically interpretable components: electrostatics, exchange-repulsion, dispersion, and induction [4013].
C70 fullerene containing a water dimer,
from [2517]
![C70 fullerene containing a water dimer [2517]](images/h2oc70.gif "C70 fullerene containing a water dimer [2517]")
Using an ingenious synthetic methodology involving opening up a fullerene, inserting the water molecule(s) under pressure and then re-sealing the fullerene, one molecule of water has been placed in C60 [2516] and C70 fullerenes [2517], and two water molecules placed inside the C70 fullerene [2517] (see right).
The water dimer inside the C70 fullerene is free from any further hydrogen-bonding to water molecules and is prevented from dissociating due to the confinement. It has a cis-linear conformation resulting from confinement (3.7 Å x 4.6 Å prolate ellipsoid cavity) and high effective pressure effects inside C70. This has a structure similar to the most energetically favorable water trans dimer (see above), with the important change in that the water molecule on the left (above) is flipped up rather than down (see right).
C84 fullerene containing a water trimer,
from [3197]
![C84 fullerene containing a water trimer, from [3197]](images/h20c84.gif "C84 fullerene containing a water trimer, from [3197]")
The cyclic water-trimer is predicted to be able to be placed into the D2(22)-C84 fullerene (one of 24 isomers) with a potential-energy gain of 43.5 kJ ˣ mol−1
Two different conformational forms are predicted to exist with the arrangement containing the three non-hydrogen-bonded H atoms on the same side of the O-O-O plane (cis-organization) slightly more stable (0.3 kJ ˣ mol -1 C84) than the one similar (but not identical) to the gas-phase form (trans-organization), as shown left (two down one up). The ratio of these two forms is about 57%:43%. The hydrogen bonds are calculated to be slightly shorter than that in the water dimer inside the C70 fullerene shown above right.
Water confined in larger fullerenes is calculated to exist in single to multiple concentric, spherical shells, as the size of the fullerene increases with these water-cluster shells exhibiting solid-like behavior at temperatures as high as 500 K [3200].
The spherical C180 fullerene can enclose the (H2O)20 dodecahedral cage, and the spherical C500 fullerene can enclose the (H2O)100 inner ES core. However, their structures are more determined by van der Waals interactions with the carbon cages than by hydrogen bonds.
The water trimer ring system has been reviewed up to 2003 [2426] and reanalyzed in 2016 [2726]. It appears that quantum delocalization of hydrogen-bonded protons between oxygen neighbors occurs in the trimer and pentamer, but not the tetramer and hexamer [2426]. Such aromatic-like delocalization within pentamers and dodecahedra, present in supercooled water, stabilize their structures and contribute to the stability of ES-clustering, where half the water molecules are within pentamers.
The average dimensions for the trimer, tetramer, and pentamer from ab initio 6-31G** calculation are shown below. The charge on the donor hydrogen atoms increases the hydrogen bond lengths contract, and the electron density width within the hydrogen bonds increases as the structure goes from dimer to trimer to tetramer to pentamer.
Structures of water trimer, tetramer, and pentamer, from ab initio 6-31G** calculation

Alternative first-principles calculations, using density functional theory (DFT) methods, of the trimer and tetramer (but not the pentamer or hexamer) reveal that the electron delocalization extends to the ring centers [3662]. The tunneling splitting patterns in some partially deuterated water trimers have been calculated [4197].
[Back to Top ![]() ]
]
(H2O)6 'cage' hexamer water cluster
with eight hydrogen bonds
6 cage hexamer water cluster")
Many theoretical studies in vacuo (e.g., [3555]) and some experimental work in the gas phase have been carried out on small water clusters. The hydrogen bond strength is unexpectedly high in these small water clusters [4110], given their non-linearity. Often the underlying theme is that the structures will be of use in understanding the structure of liquid water. This, however, is misleading as the lowest energy clusters, with greater than five water molecules, do not occur in liquid water. The reason for this is that the small water clusters described balance the strength of their hydrogen-bonding against the number of hydrogen bonds that can be formed, often maximizing the number of hydrogen bonds with each hydrogen bond being of sub-maximal strength (17-19 kJ ˣ mol−1, [2596] ) and with somewhat strained angles (poor directionality) and low entropy. If such clusters were to be immersed in liquid water, many new hydrogen bonds would be established around their periphery with the surrounding water molecules in the liquid; with the result that the original cluster would immediately 'dissolve' and change its structure. In confirmation, it has been demonstrated that the THz spectra of small to medium-size micro-solvated cations in the gas phase are fundamentally different from the bulk solution at ambient conditions [3656]. Several structures located in the solvent phase are found to be unstable
in the gas phase and vice versa [4214].
The structures of these small water clusters [3835] are interesting, however. The most stable structures with up to five water molecules are given as the above planar structures. Examples of larger clusters which have three-dimensional structures are provided right and below.
The (H2O)6 cage hexamer water cluster (shown above right) forms the structure of ice VI. They also may exist separately as stable in vaccuo clusters, with each tricyclo-hexamer resembling a distorted octahedron. By loss of the bottom water molecule, a 'tricycle' pentamer is formed. that may exist in high-density water [3462].
(H2O)8 'cube' (D2d) [2595, 4145] low energy isomer
with twelve hydrogen bonds
![(H2O)8 cube from [2595] and [4145]](images/octamer2.gif "(H2O)8 cube from [2595] and [4145]")
(H2O)6 'prism' [2533] with nine hydrogen bonds
![(H2O)6 prism from [2533]](images/prism2.gif "(H2O)6 prism from [2533]")
(H2O)6 'book' with seven hydrogen bonds
In liquid water, the preferred (H2O)6 conformation would be the chair hexamer with just six internal hydrogen bonds but with an additional twelve hydrogen bonds to other water molecules in the liquid (about twice as many and stronger hydrogen bonds than in the gas phase). Also, in liquid water, the preferred (H2O)8 conformation would be the bicyclo- octamer with just nine internal hydrogen bonds but with an additional 14 hydrogen bonds to other water molecules in the liquid (also with about twice as many and stronger hydrogen bonds than in the gas phase). Larger structures have been found by ab initio calculation (for example [115, 3780] ). As an example, (H2O)16 was shown to be optimal in a tri-stacked cube conformation with 28 hydrogen bonds; a structure that is highly unlikely to survive in a liquid water milieu, due to its tendency form more stable structure with additional hydrogen bonds to surrounding water molecules (with up to 48 intra- and inter-cluster hydrogen bonds), and that has never been found there.
[Back to Top ![]() ]
]
a The fugacity (f) of a real gas is the effective pressure which replaces the true mechanical pressure due to the non-ideality; f = φ x P where φ is the fugacity coefficient and P is the pressure. For an ideal gas f = P. [Back]
Putative water dimers

b Other putative water dimers are shown right. They do not give stable clusters using ab initio 6-31G** calculations. Six different possible water dimers may be described using a density functional theory (DFT) computational approach. The water dimer described above is the lowest energy form.
[Back]
[Back to Top ![]() ]
]
Home | Site Index | Water molecule | Water vibrations | Dimer orbitals | LSBU | Top
This page was established in 2015 and last updated by Martin Chaplin on 30 May, 2022
